Authors: PRAVEEN KUMAR*, YATER DAS AND N. CHOURASIA
ASSAM AGRICULTURAL UNIVERSITY JORHAT ASSAM-13
*Email id: bkpraveen13@gmail.com
Introduction:
NGS is referred to as massively parallel sequencing, which means that millions of small fragments of DNA can be sequenced at the same time i.e in parallel, creating a massive pool of data. Next-generation sequencing (NGS) technology, the most advanced method of genome sequencing, has become the main tool for developing novel molecular markers and high-throughput genotyping strategies, whole-genome analysis, identifying genes of agronomic importance (Edwards and Batley, 2010), constructing high density genetic maps, new experimental populations, etc. which can be incorporated into existing breeding methods It provides a golden opportunity for understanding biological systems in crops. Next generation sequencing and massively parallel DNA sequencing are blanket terms used to refer collectively to the high throughput DNA sequencing technologies available which are capable of sequencing large number of different DNA sequences in a single reaction (i.e in parallel).
NGS instruments are classified as second and third generation sequencing technologies. Second generation sequencing technology includes the 454 Roche, Illumina platforms ,SOLiD and Ion Torrent sequencers. The third generation sequencing instrument is the PacBio RS by Pacific Biosciences which does not require amplification of DNA molecules.
Before the advent of NGS technologies, sequencing by Sanger method, the time-consuming and labor intensive clone-by-clone method was used in genome sequencing with the strategy of identifying the least redundant overlapping clones. RNA-seq has shown advantages over microarrays by allowing accurate, efficient and reproducible estimations of transcript abundance of either known or unknown transcripts with a larger dynamic range using less RNA sample (Wilhelm et al. 2008; Fullwood et al. 2009).The NGS platforms, such as GS-FLX and Illumina HiSeq, become the best choice for employing the whole genome shotgun (WGS) strategy for sequencing projects of various organisms including crops because tremendous amounts of data are produced in a short period of time.
In 2002, draft genomic sequences of two rice subspecies, O. sativa ssp. japonica (Nipponbare) and O. sativa ssp. indica (93–11), were released (Yu et al. 2002; Goff et al. 2002); and subsequently, the International Rice Genome Sequencing Project (2005) completed the final genome sequence of Nipponbare. Recently, remarkable progress has been made in understanding genetic and functional diversity in rice by sequencing Oryza glaberrima (Wang et al. 2014) and 3000 other globally distributed accessions of rice (Li et al. 2014).
Advances have been made in many areas, including:
(i) Characterization of genomic variation and genetic structure in rice populations;
(ii) Analysis of the relations between genomic variations with phenotypic traits; and
(iii) Investigation of the origin of cultivated rice to further understand rice domestication and breeding history.
Commonly used sequencing method:
- Chain Termination (Sanger) method
- Illumina (Solexa) sequencing method
- Roche 454 sequencing method
- Ion torrent method
- SOLiD sequencing method
- Single Molecule real time (SMRT) sequencing by Pacific biosciences technology
General steps in NGS
i.Template preparation: The starting material for all NGS experiment is double stranded DNA. All the starting material must be converted into a library of sequencing reaction templates (sequencing library) which require common steps of fragmentation, size selection and adapter ligation, depending on sequencing platforms specification
ii. Sequencing reactions: Reactions uses a flow cell that house the immobilized templates and enables standardized addition and detection of nucleotides, washing/ removal of reagents and repetition of this cyclical process on a nucleotides by nucleotides basis to sequence all DNA templates in parallel.
iii. Data analysis: The initial analysis or base calling is typically conducted by proprietary software on the sequencing platforms. After base calling, the sequencing data are aligned to a reference genome if available or a de novo assembly is conducted.
iv. Sequencing coverage and error rates : In reality accuracy is never 100% and coverage is not uniform. Therefore, deeper sequence coverage is needed to enable correction of sequencing errors and to compensate for uneven coverage. Standards are evolving and current recommendations range from 30X to 100X coverage, depending on both platform error rate and the analytic sensitivity and specificity desired.
Sequencing methods : Second Generation Sequencing Technology :
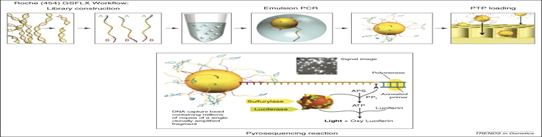
1. Roche 454 sequencing technology (Pyrosequencing) this is the first NGS released in 2005 by 454 Life Science (now Roche) and is based on principle of pyrophosphate released detection also called as sequencing by synthesis, uses luciferases to generate light for detection of the individual nucleotides output. The average read length is 200-300 bp and 80- 120 Mb per run. This technology amplifies DNA inside water droplets in an oil solution known as emulsion PCR which is based on highly efficient in vitro DNA amplification. In emulsion PCR, individual DNA fragment carrying streptavidin beads, obtained through shearing the DNA and attaching the fragment to the beads using adapter are captured into separate droplets. The droplets act as individual amplification reactors producing approx 107 clonal copies of unique DNA template per beads. A multi-enzyme complex consisting of DNA polymerase, ATP sulfurylase, luciferase and apyrase is responsible for the amplification reaction.
Each template containing beads is transferred into a well of picotitier plate. After nucleotide incorporation, the release of PPi induces the sulfurylase reaction resulting in a quantitative conversion of PPi to ATP. The luciferase uses the ATP in the conversion of luciferin to oxyluciferin. As a result of this reaction, visible light is emitted and can be detected by a CCD camera. Free ATP and nucleotides are degraded by the apyrase.
Limitations :
i. High cost of operation
ii. Lower reading accuracy in homopolymer stretches of identical bases.
iii. Noise i.e the system are not sensitive enough to detect the extension of one base at the individual DNA template molecule level.
Present scenario: The next upgrade 454 FLX Titanium will quintuple the data output from 100 Mb to about 1000Mb and the new picotitier plate in the device use smaller beads about 1 um diameter.
2. Illumina (Solexa) technology: The Illumina platform uses bridge amplification for polony generation and a sequencing by synthesis approach. Solexa was founded by S. Balasubramaniam and D. Klenemann in 1998 and sequencing was based on reversible – dye terminators technology and engineered polymerase.
All the enzymatic process and imaging steps take place in a flow cell. This method is based on a two-step mechanism and combines single molecule amplification technology and novel reversible terminator-based sequencing. First, DNA randomly fragmented by shear stress is ligated with adapters at both sides of their chain. Then the DNA is attached to the internal surface of a flow cell. This flow cell is derivatized with oligonucleotides forming a dense layer of primers, which are complementary to the adapters. The DNA fragments hybridized with the primers in a bridging way initialise solid-phase bridge amplification immediately and the fragments become double-stranded. The process is done through cyclic alteration of three specific buffers that mediate the denaturation, annealing and extension steps at 60°C resulting generation of a high-density of equal DNA fragments in a small area. Approximately one million double-stranded DNA copies are produced per cluster, which is representing one single fragment.
The second step is the real sequencing reaction. All four dNTPs carrying a base-unique fluorescent dye are added and incorporated by a DNA polymerase gradually. Following each base incorporation step, an image is made by laser excitation for each cluster. The identity of the first base is recordable. Then the elimination of the chemically blocked 3’OH group and the dye follows. Within every new cycle, the DNA chain is elongated and more images are recorded.
Limitations:
i. Short read length and hence cannot resolve short sequence reads.
ii. Due to use of modified DNA polymerases and reversible terminator, substitution error have been noted.
iii. Post phasing and prephasing decrease the signal to noise ratio per cluster, causing a decrease in quality towards the ends of the reads.
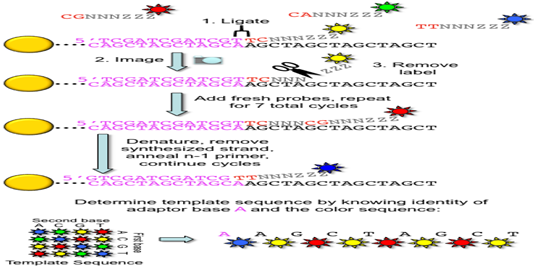
3. ABI/ SOLiD sequencing: Also called as sequencing by ligation. SOLiD is a system by Applied Bioscience DNA sequencing consist of random fragmentation of whole genome DNA and ligation of the two different 25 bp adapters (P1 and P2) at the 5’ end and 3’ ends of the DNA. In mate paired library, DNA fragment of known length are ligated such that they encompass an internal adapter. Subsequently two different DNA adapters are ligated at the 5’ and 3’ ends.
DNA fragments are ligase-modified with adapters, coupled to microparticles and applied to an emulsion PCR system. The adapters are cleaved and DNA rings are formed by ligation of the adapter ends. After the DNA accumulation steps, different fluorescence-labeled 8mer oligo - nucleotides are ligated to the sequencing primers binding to the adapter sequence. The dye color is defined by the first two of the eight bases. If the first bases fit to the DNA sequence, their fluorescence signal can be measured. After the elimination of the last three bases and the dye, the fifth, tenth and 15th base will be identified in further cycles. Other steps with shorter primers lead to the detection of the positions four, nine, 14, and so on. The process is repeated until reaching read length between 35 and 100 nucleotides. Hence, SOLiD is not running as fast as the other systems and the degradation of the template strand over time is the limiting factor.
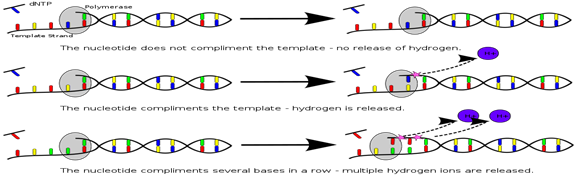
4. Ion Torrent Semi conductor Sequencing: This method is based on the detection of hydrogen ions that are released during the polymerization of DNA as opposed to the optical methods. An oil water emulsion is created to partition small reaction vesicles that each ideally contains one sphere, one library molecules and all the reagents needed for amplification. Two primers that are complementary to the sequence library adapters are present (one in the solution and other is bound to the sphere). This serves to select for the library molecules with both an A & B adapter while excluding those molecules with both adapter from loading on the beads during emPCR. This also ensures a uniform orientation of the sequence library molecules on the sphere. During the emPCR system, individual library molecules get bound to the beads to allow ultimate detection of the signal.
The Ion Torrent chip consists of a flow compartment and solid state pH sensor micro-arrayed wells that are manufactured using process built on standard CMOS technology. The detection is based on release of as H+ during extension of each nucleotide. The release of H is detected as a change in the pH within the sensor wells. Due to the lack of the time consuming image a sequence run can be completed within 4 hr. The dNTPs are added in a predefined low order. At the first release of the system, the order was a repetitive TACG sequence, later changed to more sophisticated sequence that incorporated ATAb catch up flow types to minimize de-phasing. The present error rate for substitution is ≈0.1% which is similar to that of the Illumina system. The main points of criticism in the system endures is the homopolymer errors which is 3.5%. The newest Ion 318 chips can produce ≈1 GB with an average read length from 100 upto 400bp. The proton chips I yield 60-80 million reads per run reaching 10 GB. A proton III Chip has been announced that double the number of wells to 1.2 billion leading to expected output of ≈ 64 Gb per run.
Third Generation DNA sequencing technology:
5. Single Molecule real time (SMRT) sequencing by Pacific biosciences technology :
SMRT sequencing is based on the sequencing by synthesis approach. The technology works with single molecule detection ie the optics used are sensitive enough to detect incorporation of one fluorescently labeled nucleotides. It does not require any amplification steps and the prepared library molecule is the sequencing template.
Library preparation consists of fragmentation of the gDNA to the sequence ie multi KB size, followed by end repair and either A/T overhang or direct blunt adapter ligation. After ligation the dsDNA fragments will become circular. Preparartion of the sequencing template consists of annealing a sequence primer to the ssDNA region of the SMART loop adapter, followed by binding of the DNA polymerase to form the active polymerization complex.
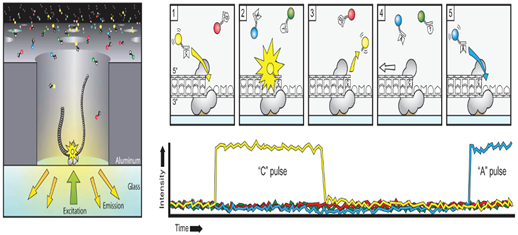
The sequencing reaction takes place at the bottom of the ≈150,000 (Zero mode wavelength) ZMWs wells on a SMRT cell. These ZMWs are small reaction wells that each ideally contains one complex consisting of template molecule, sequencing primer and DNA polymerase bound to the bottom of the ZMW. The fluorescent signals of the extended nucleotides are recorded in real time at 75 frames second for the individual ZMWs. The wells are constructed in a way that only the fluorescence occurring by the bottom of the well is detected. A powerful optical system illuminates the individual ZMWs with red and green laser beams lets from the bottom of the SMRT cell and a parallel confocal recording system to detect the signal from the florescent nucleotides.
The four nucleotides bases are labeled with a different fluorescent label and the label is detached from the nucleotides upon its incorporation into the DNA strand, leaving an unmodified DNA strand. Also, the nucleotides do not contain a terminator group allowing continuous extension of the growing DNA copy. When nucleotides complementary to the template are bound in position by the polymerase within the illumination zone of the ZMW, the identity of the nucleotide is recorded by its fluorescent label. The cleaved off label diffuses outside the illumination zone and the complex is ready for the next extension. The polymerase used for sequencing in the PacBio System is modified versions of phi 29 ie have high processivity of several hundred Kb, low error rate of ≈ 10 e -5, no GC biases and strand displacement properties. The average output per SMRT cell has increased considerably since the release of the system during 2011. The number of reads passing filter is ≈50-60 K per SMRT cell. The maxim able achievable read length can be obtained are directly related to the length of the sequencing time, but all the polymerase complexes do not provide identical read length. The main cause for this is photo-damage of the phi29 polymerase which terminates the sequencing reaction. Photo- protected nucleotide analog are used which shield the polymerase from damage and the read length of over 8-10 Kb are achieved.
With the current movie length of 180 min, read length of over 40,000 bases have been reported. In 2015, PacBio announced the launch of a new sequencing instrument called the Sequel system, with 1 million ZMWs in PacBio RS II instrument.
Comparison among the different NGS techniques:
| Method | Read length | Accuracy | Reads per run | Time per run | Cost per 1million bases (in US$) |
| Sanger | 400-900 bp | 99.9% | N/A | 20 minutes to 3 hours | $ 2400 |
| 454 | 700 bp | 99.9% | 1 million | 24 hours | $ 10 |
| Illumina | 50-300 bp | 99.9% | Up to 6 billion | 1 to 11 days | $ 0.05-$ 0.15 |
| ABI/ SOLiD | 35- 50 bp | 99.9% | 1.2-1.4 billion | 1-2 weeks | $ 0.13 |
| SMRT (PacBio) | 10,000 bp to 15,000 bp | 85.0% | 50,000 per SMRT cell | 30 minutes to 4 hours | $ 0.13 - $ 0.60 |
Limitations:
1. Requirement of high amount of 1-5 µg of total DNA or 0.5-1.0 ug sheared and size selected DNA fragments for 10Kb libraries. A lot of samples are lost during library preparation which requires stringent X P beads clean steps and Exo III and VII treatments to exclude short and/ or non- circular fragment from the library pool.
2. The high single pass error rate of 10-15 %, the majority of which are insertion / deletion errors
3. The errors is in the Pac Bio reads are randomly distributed and do not occur more frequently towards the end of the reads.
Limitations of NGS:
i. NGS offer shorter average read length (30- 400bp).
ii. During template amplification steps, mutations can be introduced.
iii. Difficult to assemble a genome de novo using short fragment lengths.
iv. Shorter read length may not align or ‘map’ back to a reference genome uniquely, often leaving repetititive regions of the genome unmappable.
v. The high volume of data generation in the range of magabases (millions) to gigabases (Millions), for which interpretations is very much trivial.
Conclusion:
The birth of NGS technologies is a landmark event in genomics, creating a new era of resequencing in a highly accelerated manner based Future goalon the high-quality reference genomes. A technology which combines the massive throughput of the NGS with the long read lengths achieved by electrophoresis-based Sanger sequencing, would enable rapid, high-quality production of de novo genome sequences. In spite of high potential, the achievements of NGS technologies have been limited to a few examples, most of which have been generated in by institutes with well established genomic facilities. A highly coordinated effort that brings together scientists and resources worldwide is a desirable step and perhaps the most efficient one for future rice genetics studies and breeding works.
Future goal
- To sequence accurately and rapidly more wild rice genomes as reference sequences
- Accelerate rice gene discovery
- Translate discoveries of natural genetic and phenotypic variations to cultivate and wild rice species.
1)Pyrosequencing method

2) illumine sequencing method:
3)AB/SoliD sequencing method:
4) Iron torrent method
5) PacBio sequencing method
Bibliography:
1.Edwards D., Batley J. Plant genome sequencing: applications for crop improvement. Plant Biotechnol. J. 2010;8:2â€"9. [PubMed]
2..https://www.illumina.com/systems/sequencing-platforms.
3.Li JY, Wang J, Zeigler RS. 2014. The 3,000 rice genomes project: new opportunities and challenges for future rice research. GigaScience 3: 1-3.
4..Wang M, Yu Y, Haberer G, Marri PR, Fan C, Goicoechea JL, Zuccolo A, Song X, Kudrna D, Ammiraju JS, Cossu RM, Maldonado C, Chen J, Lee S, Sisneros N, Baynast K, Golser W, Wissotski M, Kim W, Sanchez P, Ndjiondjop MN, Sanni K, Long M, Carney J, Panaud O, Wicker T, Machado CA, Chen M, Mayer KF, Rounsley S, Wing RA. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat Genet. 2014;46:982â€"988.
5..Goff, S.A., Ricke, D., Lan, T.H., Presting, G., Wang, R.L., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., Hadley, D., Hutchinson, D., Martin, C., Katagiri, F., Lange, B.M., Moughamer, T., Xia, Y., Budworth, P., Zhong, J.P., Miguel, T., Paszkowski, U., Zhang, S.P., Colbert, M., Sun, W.L., Chen, L.L., Cooper, B., Park, S., Wood, T.C., Mao, L., Quail, P., Wing, R., Dean, R., Yu, Y.S., Zharkikh, A., Shen, R., Sahasrabudhe, S., Thomas, A., Cannings, R., Gutin, A., Pruss, D., Reid, J., Tavtigian, S., Mitchell, J., Eldredge, G., Scholl, T., Miller, R.M., Bhatnagar, S., Adey, N., Rubano, T., Tusneem, N., Robinson, R., Feldhaus, J., Macalma, T., Oliphant, A. and Briggs, S. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science, 296, 92â€"100.
6..Yu Jin Jung, Ill Sup Nou, Yong Gu Cho, Myong Kwon Kim, Hoy-Taek Kim, Kwon Kyoo Kang, Identification of an SNP variation of elite tomato (Solanum lycopersicum L.) lines using genome resequencing analysis, Horticulture, Environment, and Biotechnology, 2016, 57, 2, 173.
References:
1. Dr. Debojit Sarma Professor At AAU Jorhat Assam Dept: Plant Breeding and Genetics
2. Dr.R.N. Sarma Professor At AAU Jorhat Assam Dept: Plant Breeding and Genetics
About Author / Additional Info:
Fellowships: ICAR-NTS , ICAR-JRf, ARS-NET, CSIR â€"NET and Ph.D. Scholar at
Jorhat Assam